Paper 6
Mode of Transcription and Maturation of Ribosomal Ribonucleic Acid in Vitro in Mitochondria from
Ehrlich Ascites Cells+
Gouder R. Kantharaj*, Kolari S. Bhat** and Narayan G. Avadhani**
National College, Bangalore, India; University of Pennsylvania,
Dept.Biology, Philadelphia , USA.
ABSTRACT: An in vitro system using mitoplasts from Ehrlich ascites mouse tumor cells was shown to be highly active in vitro protein synthesis [Bhat, N.K., Niranjan, B.G., & Avadhani N.G (1982) Biochemistry 21, 2452-2460]. In the present studies, this system was used to investigate the mode of transcription and maturation of mitochondrial 12S and 16S rRNA. The in vitro labeled RNA hybridizes to both ribosomal and nonribosomal coding sequence. The extent of hybridization to various restriction fragments suggests that the rDNA region is transcribed at 20-40 times higher RNA rates than the rest on the genome. Over 60% of the in vitro labeled RNA is adsorbed to cellulose-linked DNA restriction fragments containing rRNA coding sequences and resolves as characteristics 12S and 16S species on denaturing Agarose gels. Electrophoretic analysis of in vitro pulse – labeled RNA and Northern blot analysis of steady-state mitochondrial RNA have failed to detect significant-levels of common rRNA precursors, suggesting that the major pathway for mitochondrial rRNA maturation may involve endonucleolytic cleavage of nascent transcripts. Our results also indicate that the “D” loop area does not contribute to stable transcripts in the mouse mitochondrial system.
Mitochondria from a variety of animal cells contain a 16 kilo base pair (kbp) circular DNA genome (Borst, 1972; Dawid et al., 1976) which contains information for 2 mt rRNAs, 22 different tRNAs and potentially 13 different polypeptides (Barrell et al., 1981; Anderson et al., 1981; Bibb et al., 1981; Montoya et al., 1981: Ojala et al ., 1981) Available information in rat (Parker & Dawid, 1979), suggest that a similar organizational scheme might exist in other animal mt systems as well.
Detailed studies in HeLa cell system (Aloni & Attardi, 1971; Murphy et al., 1975) have provided evidence on the symmetrical and complete transcription of both H and L strands of mt DNA. It is also suggested that the transcription of both strands may be initiated at single promoter sites located near the origin of replication (Montoya et al., 1981; Ojala et al., 1981). These results, along with DNA and RNA sequence data (Anderson et al, 1981; Bibb et al, 1981; Montoya et al, 1981; Ojala et al, 1981) showing the absence of 3’ and 5’- untranslated regions on the mt mRNAs, and close proximal arrangement of genes with no in between spacers suggest that various RNA species such as rRNA, tRNA and mRNAs in animal cell mt are different from known pro- and eukaryotic systems.
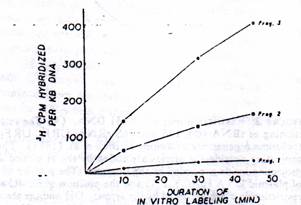
Figure 3: Rate of transcription of mt RNA and nonribosomal genes. The hybridization values from Table II were normalized for 1-kb DNA in each case (i.e, fragments 1, 2, and 3) and presented as a function of time of in vitro labeling.
Since mt RNA represents a very small fraction of total cell RNA (Avadhani et al, 1976; Lewis et al, 1976; Betty and Clayton, 1978) studies on the transcription and maturation of mt RNAs have been difficult particularly because most of the animal cells under tissue culture conditions present considerable barriers in the efficient labeling of mt transcription products.
Also, specific inhibitors like camptothecin, which have been successfully used in some cells for preferential inhibition of nuclear transcription, are not effective in other cell types (B.G Niranjan and N.G.Avadhani, unpublished results) either due to their inefficient transport across the cell membranes or due to their nonspecific effects. We have therefore attempted to develop a sub cellular system for efficient labeling of mt transcriptional products. In a previous study from this laboratory, we showed that Ehrlich ascites tumor cell mitoplasts actively incorporate [35S]-methionine into proteins resembling in vivo mt translation products (Bhat et al, 1982). In the present study we have determined the usefulness of this mitoplasts system to study mt transcription process, and as a first step towards the goal, we show that in vitro mitoplasts system is active in transcription and is capable of accurate processing of mRNA.
Experimental Procedures:
Materials: Electrophoretic grade Agarose and restriction enzymes ere purchased from Bethesda Research Laboratory. Ultra pure sucrose was from Schwarz/Mann. Sequanal grade NaDodSO4 and optical grade CsCl were purchase from Pierce chemicals Co. Poly (U- Sepharose 4B was purchased from Pharmacia. Methyl mercury hydroxide (1M solution) was from Alfa Ventron Corporation. Most of the inhibitors like Chloramphenicol, Ethidium bromide and Cordecepin triphosphate were purchased from sigma chemicals Co. [3H] CTP (18 Ci/mmol), and [32P] dCTP (>3000 Ci/mmol) were obtained Amersham Corp. Nitrocellulose membrane sheets for RNA blot transfer were purchased from Schleicher & Schuell.
Isolation of Mitochondria: Ehrlich ascites mouse tumor cells were grown in the peritoneal cavity of Swiss mice (Chun et al, 1969) and used as the source of mitochondria. Disruption of cells by homogenization in sucrose-mannitol buffer (4mM Hepes, pH 7.4, 220mM mannitol, 70mM sucrose, and 2mM EDTA) and isolation of crude mitochondria by differential centrifugation were as described before (Lewis et al, 1976, Niranjan and Avadhani, 1980; Bhat et al, 1981). Mitochondria were washed once with sucrose-mannitol buffer containing 20mM EDTA, and the mitoplasts were isolated by the digitonin (fractionation method using 0.1 mg of digitonin/mg of mt protein, also as described before (Bhat et al, 1982).
In vitro labeling of mitoplasts and isolation of mt RNA: mt RNA was labeled with 3H-labeled or32P-labeled nucleotides by using an in vitro system previously described for labeling mt translation products (Bhat et al, 1981, 1982). Freshly prepared mitoplasts were suspended in the RNA synthesis buffer [5mMhepes, pH7.4, 60mM KCl, 6mM Mg (CH3-COO) 2, 5 mM 2-mercaptoethanol, 3 mM KH2PO4, pH 7.4 and 0.14 M sucrose] at a concentration of 6-10 mg of mitoplast protein/ml and supplemented with 2mM ATP, 1 mM GT, 5mM creatine phosphate, 4 mM pyruvate, 0.2 mg/ml creatine phosphokinase, and 100 uM each of 20 amino acids. The mixture was gently shaken at 35^oC, and the labeling was initiated by adding 100 uCi/ml each of [3H] CTP (18 Ci/mmol), and [3H] UTP (50uCi/mmol) or 100 uCi/ml [32p] UTP (>600 Ci/mmol). Unless otherwise stated, labeling was continued for 60 minutes. Aliquots (2.5 ul) were withdrawn at intervals, adsorbed onto filter discs, and assayed for radioactive RNA synthesis by cold Ccl3COOH method (Mans & Novell, 1961).
The labeled mitoplasts were pelleted at 10 000g for 10 min at 4oC, washed once with sucrose-mannitol buffer, and used for isolation of mt RNA by phenol-chloroform method (Avadhani, 1979) with the following modifications. The mt pellet (5-10mg) was suspended in 2.5ml of guanidinium thiocyanate buffer (25mM sodium citrate, pH7.0, 5M guanidinium thiocyanate, 0.1M 2-mecrcaptoethanol, and 0.5% sodium laurylsarcocinate) by homogenization with Dounce homogenizer (Chirgwin et al, 1979). The clear lysate was extracted with equal volumes of phenol saturated with water and chloroform. The aqueous phase was separated and saved, and the interphase was resuspended in guanidinium thiocyanate buffers above and extracted again with phenol-chloroform. The combined aqueous phases were rextracted once with phenol-chloroform and once with CHCl3-isoamyl alcohol (95:5) and adjusted to pH 5.0 by adding 0.025 volume of1 MCH3COOH. RNA was precipitated with 2 volumes of ethanol in the presence of 0.3M CH3COOK (pH 5.0). RNA was further pelleted through CsCl (Chirgwin et al, 1979) to eliminate contaminating DNA.
Preparation of Plasmid DNA: E.coli C600r-m- transformed with pACYC 177 plasmids carrying the entire mouse mt genome (designated as pAM1) was a gift from Dr. David A.Clayton (Martens & Clayton, 1979). The growth of cells in PO4 L broth and lyses of cells with Lysozyme-EDT and triton X-100 were according to Battey & Clayton (1978). The lysate was clarified at 75000g for 30 min at 4oC and was made to 10% with polyethylene glycol). The nucleic acid precipitate was collected by centrifugation at 25000g for 30 min at 4oC, digested with pancreatic RNase (DNase free), and used for the isolation of closed DNA by CsCl banding (Clewell & Helinski, 1970).
Isolation of Nick Translation of Restriction Fragments: The plasmid DNA containing mouse mt genome (10-50ug) was digested to completion with EcoR1 and Bgl I under standard conditions recommended by the vendor. The DNA fragments were resolved by electrophoresis on 0.6% Agarose slabs and localized by staining with 0.5ug/ml EtBr. The DNA from the gel slices was electro blotted on DEAE paper (NA 45, Schleicher & Schuell), eluted by extraction with 1M NaCl, and precipitated with 2.5 vol of ethanol. The DNA fragments were Nick translated with [32P]-dCTP (>3000Ci/mmol) by using a kit supplied by Bethesda research laboratory.
Covalent Binding of DNA to Cellulose: About 200-250 g of mt DNA (restricted fragments) in 1ml of H2O was solicited for 10s and denatured by heating at 100oC, followed by quick chilling in ice. The single stranded DNA fragments were covalently linked to epoxy-cellulose using the method of Moss et al, (1981).
Electrophoretic Analysis of RNA: RNA was separated on denaturing Agarose methyl-mercury hydroxide gels (Baily & Davidson, 1976).
After the electrophoresis run, the gels were stained with 1ug/ml ethidium bromide in 0.5m NH4CH3Coo, and the RNA bands were visualized under UV light. 3H-labeled RNA bands localized by fluorography by using EN3HANCE (New England Nuclear). In northern blotted experiments (Alwine et al, 1977), 1977, NA from the gels was blotted onto nitrocellulose sheets as described by Thomas (1980) and probed with 32P-labeled DNA fragments (Alwine et al, 1977).
DNA-RNA hybridization: DNA (2-10ug) in 100ul of 0.3M NaCl and 0.3M NaOH was denatured by heating at 100oC for 15 min following quick chilling in ice. DNA was spotted on nitrocellulose disks and immobilized by heating at 80oC under vacuum for 2h. Hybridization was carried out in 0.5-1 ml of reaction mixture containing 5 x SSPE9 (1XSSPE=0.18M NaCl, 10mM sodium phosphate, pH 7.1, and 1mM EDTA), 50% formamide, 2 2x Denhardt’s solution [0.04% each of Ficoll, poly (venylpyrrolidone), and bovine serum albumin], 50ug/ml yeast RNA at 42oC for 48hr. The filters were washed with 25ml of 2x SSPE and 25ml of 1x SSPE. The filters were digested with 50 ug of pancreatic RNase in 1ml of 1x SSPE, washed with 10ml of 1x SSPE, air dried, and counted with 10ml of ACS II scintillation mixture Amersham).

Figure 1: Mitoplasts from Ehrlich ascites cells isolated by digitonin fractionation were labeled with 1(x) uCi/mL [3h]UTP and [3H]CTP as described under experimental procedures. Aliquots (2.5ul) were assayed for cold CCl3COOH insoluble cpm. Ethidium bromide when added* was at 2ug/ml, and 3’ATP when added was at 25 ug/ml level (o).

Table 1: Mitoplasts from Ehrlich ascites tumor cells were labeled with 100 mCi/mL each [3H] UTP and [3H] CTP as described under Experimental Procedures. At the end of labeling, mitoplasts (7.5) were pelleted at 10000g for 10 min and washed once with sucrose – mannitol buffer. The pellets were dispersed in 2.5 mL each of guanidinium thiocyanate buffer and RNA was extracted as described under Experimental Procedures. RNA was estimated by using an extinction bromide and 3’-dATP were added at zero time to the concentrations of 2 mg/mL and 25 mg/mL, respectively.
Results:
RNA Synthesis in Isolated Mitoplasts: In a recent study from this laboratory, it was shown that digitonin-treated mt particles from LES cells are highly active in [35S] methionine incorporation (Bhat et al., 1981, 1982). Essentially, the same incubation system was used to label total mt RNA and study the synthesis and maturation of mt rRNA. The rate of incorporation of [3H] CTP and [3H] UTP by LES mitoplasts in presented in Figure 1. Ethidium bromide at 2 ug / ml inhibits the incorporation by about 75% and 3’dATP at 25ug/ml inhibits the incorporation by about 60%. Although not shown here, the rate of inhibition by both ethidium bromide and 3’dATP are dose dependent. The specific activity of mt RNA labeled in vitro with [3H] CTP and [2H] UTP for various time intervals has been presented in Table 1. It is seen that the specific activity increases from about 2500 cpm/ug of RNA at 15 min of labeling to about 6200 cpm/ug of RNA at 45 min of incubation, suggesting a steady accumulation of newly synthesized RNA. As shown for the total acid precipitable cpm above, addition of 2ug/ml EtBr or 25 ug/ml, 3’-dATP results in 60-7-% reduction in the specific activity of mtRNA. A nearly 60% higher specific activity is obtained with 200uCi/ml each of [3H] UTP and [3H] CTP. Also use of 250 uCi/ml each of [32P] UTP and [32P] CTP yield about 3.5 x 10^4 cpm/ug of mtRNA (results not shown).
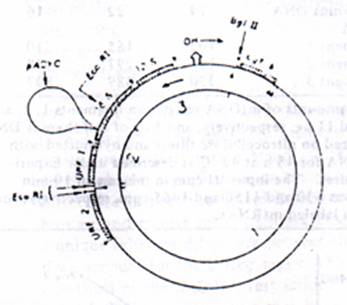
Figure 2: Restriction map of pACYC M1 DNA. (A) The relative positioning of tRNA (O); 12S and 16S rRNA; URF1; URF3 and cytochrome b genes are according to Bibb et al, (1981). The tRNAs Projecting outside the circles are coded by the H strand and those projecting inside are coded by the L strand. The position of insertion of plasmid DNA (pYCYC-177) and the position of insertion of E.coR1 and Bgl ii restriction sites are indicated by the arrows. OH (origin-H) and the arrow outside the circle indicate the origin and direction of H strand DNA synthesis. The arrows inside the circle indicate the direction of H Strand transcription. 9B). The pAM1 DNA was digested to completion with Bgl lII and E.co RI and electrophoresis on 0.6% Agarose gels, and the DNA bands were visualized under UV light after staining with EtBr as described under experimental procedures.
Nature of Mitochondrial DNA probes Used. Hybridization of in vitro labeled mt RNA to mt DNA restriction fragments containing rRNA genes and various nonribosomal genes was used to determine the extent of transcription of mt genome and the nature of RNA synthesized under the in vitro conditions. In these studies, the mouse from LA 9 cells cloned in PACYC-177 plasmid vector (Martens & Clayton, 1979) was used to prepare the specific probes. The restriction maps for Bgl II and Eco RI on the cloned mt genome are shown by the arrows in the figure 2. The relative positioning of 12S and 16S rRNA, the tRNAs and presumptive mRNA cistrons on the mt genome, as shown in figure 2, was based on the DNA sequence data reported by Bibb et al, (1981). The point of insertion of PACYC plasmid at unique Hae II site on the mt Genome lies about 70 nucleotides from the 3’ end of the 16S rRNA coding sequence (see figure 2). Double digestion of cloned DNA with Bgl II and E co RI yields three large DNA fragments as shown in Figure2 and one small fragment of 200 nucleotides which migrates out of the gel under the Electrophoretic conditions used. Following the terminology of Bibb et al. (1981), the largest fragment of 11.3 kb DNA contains 12 different reading frames potentially coding for mRNA and 15 different tRNAs. The second largest fragment of 5.7 kb contains URF1 gene, 0.5 of the 16S rRNA gene (925 nucleotides), 3 tRNA genes, and 3.7 kb plasmid DNA. The third fragment of 2.7 kb contains the 12S rRNA gene, 0.5kb of 16SrRNA gene (725 nucleotide), 3 tRNA genes, and all of the D-loop area. For the sake of presentation, these three DNA restriction fragments will be referred to as fragments 1, 2, and 3, respectively as indicated in Figure 2B.

Table 2: Equimolar amounts of mt RNA restriction fragments 1,2 and 3 i.e. and 11 ug respectively, and 5ug of each calf thymus DNA were immobilized on nitrocellulose filters and hybridized with 0.3 ug of mt RNA for 43hrs at 42^C as described under experimental procedures. The input in the case of 10-mi labeled RNA was 630, and 1150 and 1465 cp, respectively, for 30 and 45 min labeled mt RNAs.
Extent of Transcription of mt Genome: The extent of transcription of mt genome under the in vitro conditions was determined by hybridizing in vitro labeled mtRNA to the three mt DNA restriction fragment described above (Table II).
Fig 3: Rate of transcription of mt rRNA and nonribosomal genes. The hybridization values from table II were normalized for 1-kb DNA in Each case (i.e, fragments 1, 2 and 3) and presented as a function of time of in vitro labeling.
It is seen that RNA labeled for a short duration of 10 min as well as those labeled for 45 min hybridize significantly to all three mt DNA fragments, suggesting that genomic area coding for both rRNA an mRNA ARE transcribed under the in vitro conditions. The rate of transcription of different genomic areas appear to vary considerably (Table II) since nearly 60% of the counts hybridize to fragment 3 which contain rRNA genes, whereas only 10-15% hybridize to fragment I which mainly contains the mRNA and tRNA genes. Although about 25% of the labeled RNAs hybridize to mt DNA fragment 2, it should be noted that this contains a portion of the 16s rRNA coding sequence n addition to genes coding for URF1 and two tRNAs. Further, since there is a vast difference ( about 5-fold) in the size between fragment 3 and fragment I, the hybridization results in Table II were normalized for a unit length of DNA (1kb) and presented in the figure 3. These results clearly show that at 10, 30, and 45 min of in vitro RNA labeling, the rRNA region is transcribed 20-40 times faster than the rest of the genome. These results are in agreement with the published reports showing a considerably faster rate of metabolic labeling of mt rRNAs as compare to that of nonribosomal transcripts in HeLa cells (Gelfand & Attardi 1981; Attardi et al 1982) and 20-60 fold abundance of mt 12S and 16S rRNA in the steady-state mt RNA (Battery & Clayton, 1978).
Mode of Synthesis of mt rRNA. In order to study the made of synthesis of mt rRNA and also to identify putative precursors of rRNAs pulse – labeled for 10, 30 and 45 min were electrophoresed on 1.6% Agarose – methyl mercury gels. As shown in the Fig 4, all of the three [3H] RNA sizes contain two major components with Electrophoretic mobility characteristic of mt 12S and 16S rRNAs. These two RNA bands together account for over 60% of the total input label, providing support to the hybridization results which showed that rRNA genes are transcribed at 20-40fold higher frequency than the rest of the genome.
A B C

Figure 4: Electrophoretic pattern of in vitro labeled mt RNAs. Mitochondrial RNA was labeled in vitro using 100uCi/ml of [3H] and [3H] CTP as described in Figure1 and Table 1, for 10, 30, and 45 min. A. 10ug sample of RNA in each case electrophoresed on 1.6% Agarose methyl-mercury gels. Similarly 20ug of 10-min labeled [3H] RNA was adsorbed to cellulose bound DNA restriction fragment 3 (about 100ug of DNA) and a specifically hybridized was eluted as described under experimental procedures and electrophoresed as above. The gels were fluorogrammed by using ENHNCE, (lane 1) mt RNA labeled for 10 mi, 2) mt RNA labeled for 30 min; (lane 3) mt RNA labeled for 45 min; (lane 4), 10-min labeled RNA hybridized to cellulose bound restriction fragment 3.
Studies on HeLa cell mt transcription (Gelfand & Attardi, 1981); Ojala et al, 1982) suggested the occurrence of low abundance poly (A) RNA having the properties of a common precursor to both 12S and 16S rRNAs. The in vitro 3H-labeled RNA, however, does not appear to contain detectable levels of putative mt rRNA precursors during a short pulse labeling of 10 min (Figure 4, lane 1-3). Ribosomal nature of two RNA bands shown in Figure 4 (lanes 1-3) was ascertained by specific hybridization of cellulose bond mt DNA.
As shown in Fig.4 (see lanes 4) 1min in vitro labeled RNA hybridized to cellulose –bound mt DNA restriction fragments 3 shows the presence f 12S and 16S RNA with no detectable precursor species. In a separate experiment involving high specificity activity labeling with [32P] UTP, although the synthesis of some high molecular weight RNA in the size range of 2.5-13.6 kb is noticed. The high molecular weight mtRNA do not hybridize to cellulose bound DNA fragments (data not presented), suggest their nonribosomal nature.
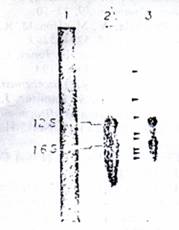
Figure 5: analysis of steady-state mt RNA by northern blot. Total mtRNA (10ug) isolated from ETA washed mitoplasts as described under experimental procedures was electrophoresed on 1.6% Agarose-methyl mercury gels. The RNA was blot transferred to nitrocellulose papers and probed with nick-translated mt DNA restriction fragments 6.2 x 10^8 32P cpm) as described under experimental procedures (Lane 1). Ethidium bromide stained pattern of mt RNA; (Lane 2) mt RN probed with nick-translated DNA fragment 3; (Lane 3) mt RNA probed with nick-translated pAM1 DNA. Arrows in Lane 3 indicate the presumptive mRNAs and pre-mRNAs.
The possible occurrence of putative RNA precursors in the steady state mt RNA was determined by using RNA blot transfer experiments (Alwine et al, 1977). As seen in Figure 5, the EtBr-stained pattern of mt total RNA shows the presence of two major rRNA bands and a number of minor RNA species smaller than 16s rRNA. When the RNA blots are probed with 32P-labeled nick translated mt DNA fragments 3, two major bands corresponding to 16s and 12S rRNAs are seen (Figure, 5, lane 2). Even five times longer exposure of blot to X-ray films failed to identify RNA species larger than the RNA by this probe (results not shown). Use of nick-translated total Amt NA, however, shows the presence of larger RNA species 92.8kb and 3.6kb), presumptive mt precursor in addition to a number of putative mRNA species as shown for HeLa mt RNAs (Gelfand & Attardi, 1981; Montoya et al, 1981). These results using specific mt DNA probes show the absence of detectable rRNA precursors in steady state mtRNA and also in mt RNA synthesized in vitro in isolated mt particles.
Discussions:
In a previous paper from this laboratory, it was shown that Ehrlich ascites tumor cells mt can synthesize heterogeneous size class RNA, which is found to be associated with ribo-nucleoprotein particles resembling polysomal structures with to sedimentation property (Avadhani et al, 1974).
Similarly several investigators have shown that isolated mt particles from Ehrlich ascites as well as from rat liver can synthesize poly (A)-containing RN (Ajme et al, 1974 & Freeman, 1976; Rose et al, 1975; Cantatore et al, 1976). To our knowledge, however, the accuracy of transcription and also the exact nature of RNA synthesized in these in vitro systems from mammalian cells remain unclear. Recently it was shown that the yeast mt particles are able to synthesize putative precursors of 21S and 15S rRNAs under in vitro conditions (Boerner et al, 1981; Groot et al, 1981). The labeled pre 21S rRNA in the mt particles was converted to mature 21S rRNA, a process which includes “splicing” of 1.1 kb intervening sequence and trimming at 3’ends. The processing of pre 15.5S rRNA to mature 15S rRNA was, however, restricted in this in vitro system (Boerner et al, 1981; Groot et al, 1981). In this paper we report on the qualitative nature of RNA synthesized in Ehrlich ascites mt in vitro and show that both the 16S and 12S rRNAs are synthesized with accuracy n this system.
Hybridization of in vitro labeled RNA to mt DNA restriction fragments 1, 2, and 3 shows that both ribosomal and nonribosomal regions of mt genome are transcribed in the isolated mt particles (Table II). Further digestion of DNA fragment 1 (11.3kb) with Hpa I and Hind III yields six restriction fragments of 0.9-3.8 kb, and the in vitro labeled mt RNA hybridizes to al these six fragments (results not presented), suggesting the compete transcription of the genome under these conditions. The rate of hybridization of in vitro labeled RNA to different fragment varies markedly (Table II). Data on the DNA excess hybridization presented in Table II and figure 3 shows that on a unit basis, the rRNA region may be transcribed >30-fold higher rates than the rate of the genome.. Moreover, restriction fragment 3 (2.kb) containing the tRNA genes also contains a 750-nucleotide D-loop area (see figure 2). In human mt DNA this region is actively transcribed into an abundant poly (A)-containing of unknown function, designated as 7S RNA (Attardi et al, 1982). In confirmation with the results of Van Etten et al, (1982), however, we have been unable to detect the 27S RNA or its counterpart in the steady state mt RNA (Figure 5). Although the significance or reasons for this apparent difference in the stability of D-loop transcript between the human and mouse mt system are unknown, it may indicate a unique inherent difference between because these two systems, thus if we account for the D-loop region which corresponds to about one third of the mt DNA restriction fragment 3, the relative rate of rDNA transcription may be over 40 times that of the rest of the genome. These results are in general agreement with higher rate of labeling of mt rRNA in vivo and the relative abundance of rRNA in steady-state mt RNA (Gelfand & Attardi et al, 1982; Bettey & Clayton, 1978; Bibb et al, 1981; Dubin et al, 1982).
Transcription of H-strand is believed to be initiated at a single promoter located adjacent to the D-loop area (Attardi et al, 1980, Eperon et al, 1981), and the chain elongation appears to continue until the opposite end of the molecule (Murphy et al, 1975). Therefore, the observed 20-40 fold higher rate of transcription of rDNA in the present study and also in previously reported instances (Battey & Clayton, 1978; Gelfand & Attardi, 1981; Attardi et al, 1982) raises several possibilities. First preferential transcription of rDNA may be accomplished by chain termination at the end of 16S rRNA gene by a mechanism similar to that of bacterial transcription- attenuation (Attardi et al, 1980; Van Etten et al, 1980). In fact, sequences reminiscent of hairpin oligo (U)-structures believed to be the stop signal for E.coli polymerase (Rosenberg & courts, 19790 have been reported to occur near the 3’ end of human, mouse, ovine and hamster mt 16S rRNA (Eperon et al, 1980;Van Etten et al, 1980; Dubin et al, 1981, 1982). The second possibility is the involvement of two distinct RNA polymerases, one for the transcription of the entire genome and the second for the preferential transcription rDNA. As yet, however, there is no indication for the occurrence of more than one type of mt-specific RNA polymerase. Thus, the precise mechanism regulating this differential expression of H-strand remains to be determined.
The occurrence of low abundance poly (A)-containing RNA in HeLa mt RNA which hybridizes to 12S and 16S coding region on the mt DNA Ojala et al, 1980) suggest the possibility that the 12s and 16S mt RNAs may be derived from a common precursor. In the present study, our attempts to detect such putative rRNA precursors either in the in vitro labeled mt RNA or in steady state RNA have been unsuccessful. First, in vitro RNA specifically hybridized to cellulose-linked restriction fragment 3 consists of only 12S and 16S RNA species with no detectable precursor even in 10-min pulse labeled RNA (figure 4). Similarly, analysis of the steady-state mt RNA by Northern blot with 32P-labeled nick-translated mt DNA fragment 3 probe does not show the presence of rDNA transcripts larger than 16S rRNA (figure 5). Use of mt DNA fragment I or total mt genome as a probe, however, detects the presence of 2.4-3.8 kb transcripts, possibly representing the mt precursors. Because of the limitations of hybridization conditions, however, we cannot completely rule out the presence of such precursor. It is estimated that such a precursor, if present may occur at concentration less than 1 n 500 molecules of rRNA. Our results, therefore, demonstrate that the major pathway for rRNA maturation in mouse mitochondria does not involve the synthesis of a common precursor of 12S and 16S rRNA. This mode of maturation is consistent with the results reported for HeLa mt (Gelfand & Attardi, 1981). In conclusion RNA synthesis in this in vitro system appears to resemble in vivo mt transcription with respect to the rate of transcription and mode of RNA maturation.
Acknowledgements:
We are thankful to Dr. David A. Clayton for a gift of cloned mt DNA used in these experiments. We are also thankful to John Cozza Fluellen for excellent technical help and to Nina Leinwand for her help during preparation of the manuscript.
References:
Aloni, Y,& Attardi, G (1971) J.Mol. Biol, 55, 251-267.